Congratulations!
Kai Zhang's paper on a fast, scalable and versatile tool for analysis of single-cell omics data is published in Nature Methods!
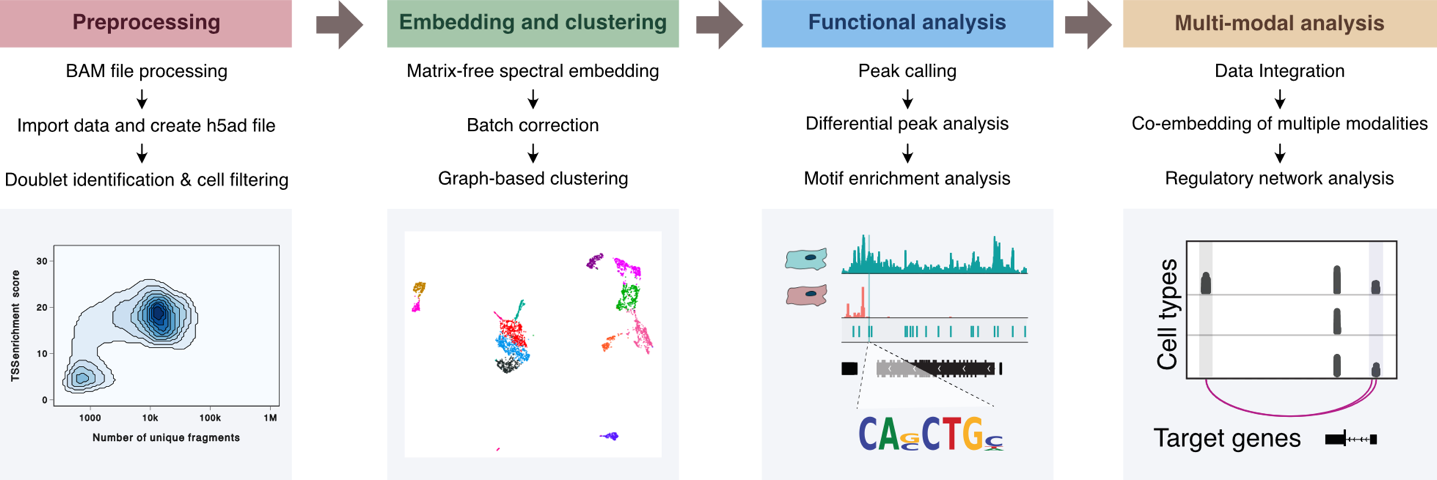
Abstract: Single-cell omics technologies have revolutionized the study of gene regulation in complex tissues. A major computational challenge in analyzing these datasets is to project the large-scale and high dimensional data into low-dimensional space while retaining the relative relationships between cells in order to decompose the cellular heterogeneity and reconstruct cell-type-specific gene regulatory programs. Traditional dimensionality reduction techniques, however, face challenges in computational efficiency and in comprehensively addressing cellular diversity across varied molecular modalities. Here we introduce a nonlinear dimensionality reduction algorithm, embodied in the Python package SnapATAC2, which not only achieves a more precise capture of single-cell omics data heterogeneities but also ensures efficient runtime and memory usage, scaling linearly with the number of cells. Our algorithm demonstrates exceptional performance, scalability, and versatility across diverse single-cell omics datasets, including single-cell ATAC-seq, single-cell RNA-seq, single-cell Hi-C, and single-cell multiomics datasets, underscoring its utility in advancing single-cell analysis.
Full text can be accessed from the following link